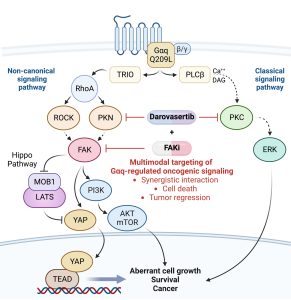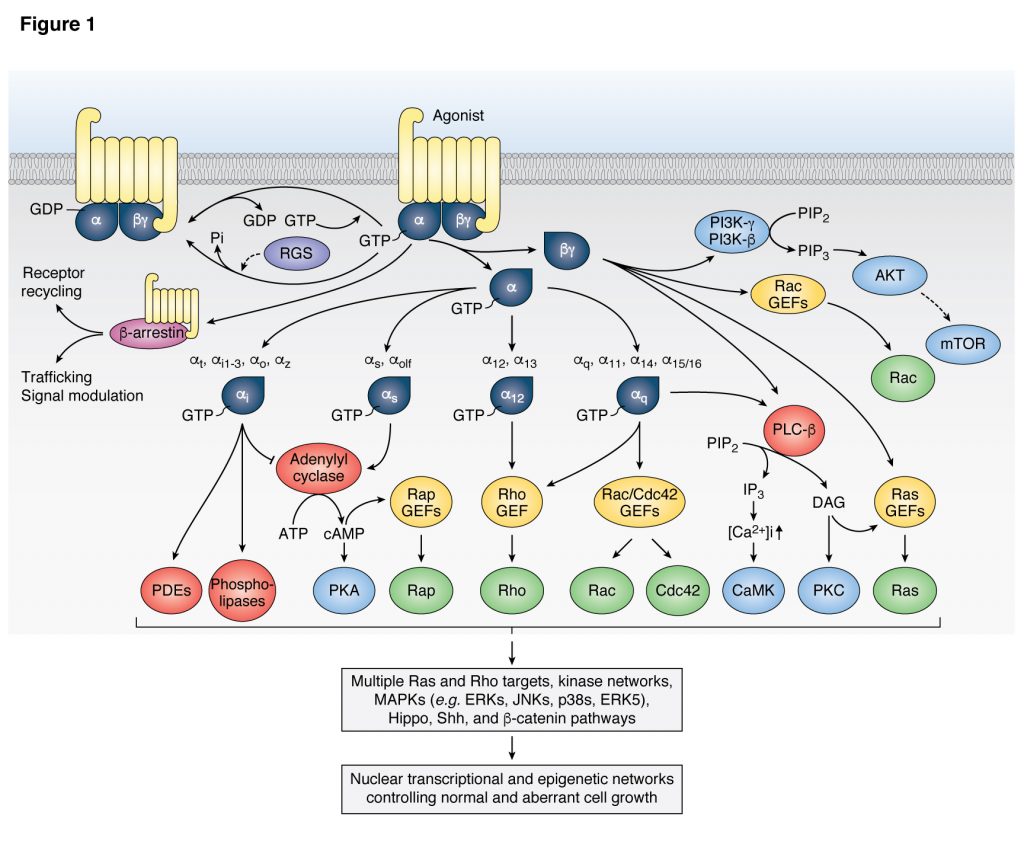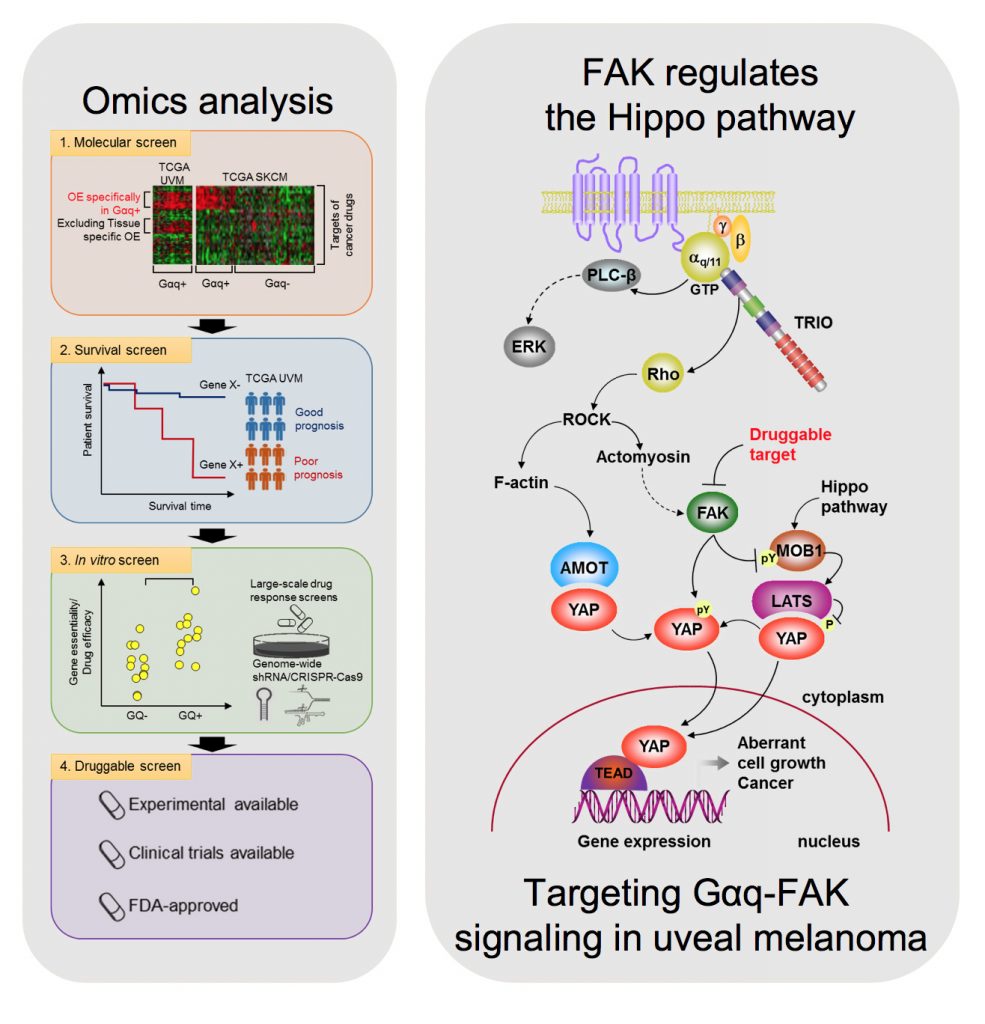
Uveal melanoma (UM) is the most common primary cancer of the eye in adults, and is the second most common melanoma subtype after skin cutaneous melanoma (SKCM). UM is diagnosed in about 2,500 patients each year in the United States alone, nearly 50% of which will die from liver metastasis within 5-10 years after diagnosis, independent of the successful treatment of the primary lesions. Activating mutations in GNAQ and GNA11 (often referred to as GNAQ oncogenes, which encode GTPase deficient and constitutively active Gαq proteins), were identified in ~93% of UM and 4% of SKCM, respectively, where they act as oncogenic driver oncogenes. These findings and our team’s early discovery that activated mutants of Gαq represent a new class of oncogenes firmly established UM and a subset of SKCM as Gαq-driven human malignancies. Indeed, this provides a clear example of a human malignancy that is initiated by gain of function mutations in Gαq and Gα11 proteins. The best-known downstream signaling event initiated by Gαq involves its ability is to activate phospholipase C (PLC) β and the consequent increased hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to produce two second messengers: inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 raises cytoplasmic Ca2+ levels, which stimulates multiple calcium-regulated pathways and, together with DAG, activates classic protein kinase C (PKC) isoforms. However, the molecular events underlying GNAQ-driven malignancies are not yet defined, thus limiting the ability to develop novel anticancer-targeted therapies. In our recent studies, we found that activating mutation of Gαq can trigger YAP translocation into the nucleus and stimulates YAP-dependent transcription, and that this process is independent from PLCβ stimulation but requires the activation of a Gαq-regulated guanine nucleotide exchange factor, TRIO, and the subsequent activation of the small GTPases RhoA and Rac1 and their associated signaling networks. Gαq activation is sufficient to stimulate YAP in animal cancer models, and knock down of Gαq in uveal melanomas harboring GNAQ mutations reduces the nuclear location of YAP, and YAP-dependent gene transcription. Gαq-TRIO-Rho/Rac signaling circuitry contributes to YAP-dependent growth in uveal melanoma, which is the first described GNAQ/GNA11-initiated human malignancy, and, thus, that YAP may represent a novel therapeutic target for uveal melanoma treatment.
However, there are currently limited option to target YAP therapeutically. Thus, there is an urgent need to understand how Gαq promotes cancer growth in order to develop new targeted (precision) therapeutic options. In a recent study, we used a novel computational framework to shed light on Gαq biology and identify systems vulnerabilities for UM, based on the prediction of synthetic (dosage) lethal gene interactions of Gαq. This novel pipeline integrates data from large multiomics cancer datasets, including UM patient transcriptomes and genomes, with in vitro screens (datasets of gene essentiality and dependence and druggable screens (datasets of drug response screens). The top predicted synthetic lethal gene with GNAQ was PTK2, suggesting the potential benefit of targeting the PTK2 gene product (a non-receptor tyrosine kinase known as FAK) in UM. Remarkably, activation of FAK by Gq-linked receptors was initially reported by our team in the early 90s. This unexpected convergence of computational predictions, biochemical, and genetic information prompted us to focus on the role of this Gαq-tyrosine kinase signaling axis in UM. We found that gene editing or inhibition of FAK reduces UM cell growth and tumor formation in vivo. Furthermore, systematic dissection of the underlying mechanisms led us to uncover that FAK promotes YAP activity through direct tyrosine phosphorylation of YAP concomitant with the release of inhibitory core Hippo signaling on YAP. Our studies provided a novel direct link between Gαq-FAK driven tyrosine phosphorylation networks and YAP activation. Based on these findings, the use of FAKi for the treatment of metastatic UM is under current evaluation, as a single agent and as part of a signal-transduction based multimodal therapy.
As part of these studies, we have used unbiased genetic screens to identify new treatment options for UM. We discovered that inhibition of the non-receptor tyrosine kinase FAK and the adaptive activation of the ERK pathway downstream from GNAQ results in UM cell death and regression of liver metastatic lesions. This provided the foundation for the first signal transduction-based multimodal precision therapy in metastatic UM (mUM)(NCT04720417). Our recent large chemogenetic drug screen revealed that targeting FAK and PKC/PKN represents an effective precision-guided combination, which we plan to explore in the clinic shortly.